EKG bei hypertropher Kardiomyopathie (HCM)
Die hypertrophe Kardiomyopathie (HCM) ist charakterisiert durch eine vorwiegend septumbetonte Hypertrophie des linken Ventrikels. Sie ist in vielen Fällen eine monogenetisch bedingte Erkrankung, bei der Mutationen in Genen des kontraktilen Apparats des Myokards zu abnormalen Strukturveränderungen führen.In etwa 60–70% der Fälle liegt eine Obstruktion des linksventrikulären Ausflusstrakts (LVOT) vor – entweder in Ruhe oder erst unter Belastung (dynamische Obstruktion).In diesen Fällen wird von einer hypertrophen obstruktiven Kardiomyopathie (HOCM) gesprochen. Eine apikal betonte Hypertrophie ist eine seltenere Variante mit charakteristischem EKG-Muster.
Die klassische Trias der HCM-EKG-Zeichen umfasst:
- Hypervoltage und LVH-Kriterien – Spannungserhöhung in den Brustwandableitungen, positive Hypertrophie-Indizes (z. B. Sokolow-Lyon-Index >3,5 mV oder Cornell-Voltage).
- Tief präterminal negative T-Wellen – besonders in den linkspräkordialen Ableitungen (V4–V6), manchmal auch in Brustwandableitungen V1–V3; charakteristisch für die abnormale Repolarisierung des hypertrophierten Ventrikels.
- Pseudo-Infarkt-Q-Zacken – tiefnegative, breite Q-Wellen in mehreren Ableitungen (z. B. II, III, aVF, V1–V3), verursacht durch die abnorme Erregungsausbreitung im septumbetonten Myokard, nicht durch Myokardinfarkt.
Diagnostische EKG-Veränderungen
1. Hypervoltage und Hypertrophie-Zeichen
- Sokolow-Lyon-Index: S in V1 + R in V5/V6 >3,5 mV
- Cornell-Voltage: RaVL + S in V3 >2,0 mV (Frauen) oder >2,8 mV (Männer)
- Breite QRS-Komplexe: Oft >100 ms, besonders bei medialer oder apikaler Hypertrophie
2. Tiefnegative T-Wellen
- Verteilung: V1–V6, I, aVL; besonders prägend in V4–V6
- Konfiguration: Präterminal negativ (breites, tiefes Negativum), nicht spitzwinklig wie bei Ischämie
- Amplitude: Oft >10 mm Negativität
- Dynamik: Kann sich im Zeitverlauf verändern; teilweise reversibel unter Belastung
Spitzwinklige, maximal tiefnegative T-Wellen in V4–V6 – oft symmetrisch und mehr als 10 mm tief sidn pathognomonisch für die apikale Form der HCM (aHCM).
3. Pseudo-Infarkt-Q-Zacken
- Breite Q-Zacken (≥40 ms Breite) in mehreren Ableitungen
- Tiefnegativ: Oft >25% der R-Welle Amplitude
- Lokalisationen: V1–V3 (verbreitet), II, III, aVF (inferiore Region), manchmal auch anterolateral
- Ursache: Abnorme linksventrikuläre Erregungsausbreitung, nicht durch Myokardinfarkt
4. Weitere Veränderungen
- Linksabweichung der QRS-Achse: Häufig bei HCM (bis zu 50% der Fälle) - Ursache: Veränderte Erregungsausbreitung durch Septumhypertrophie und myocardial disarray.
- Linksschenkelblock: ein vollständiger LSB ist eher selten (ca. 10%), wenn vorhanden, suggeriert er eine fortgeschrittene Erkrankung oder eine frühere Intervention (z. B. Septumablation). Partielle LSB-ähnliche Muster sind häufiger
- ST-Strecken-Veränderungen: subtile ST-Senkungen in V1–V3 möglich, ST-Hebungen in V1–V2 sind kein Hinweis auf akuten Infarkt, sondern spiegelbildliche Veränderungen oder Repolarisierungsvarianten.
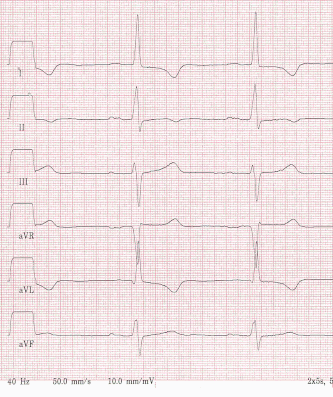

Abb.: 59-jähriger Mann mit HCM. Die EKG-Veränderungen sind pathognomonisch. In V1 zeigt sich eine „Pseudo-Infarkt-Q-Zacke“. Die Obstruktion lag midventrikulär. Bei der apikalen Form der HCM würde man eine maxiamel Ausprägung der tiefnegativen T-Wellen in V5 und V6 erwarten.

Abb.: 55-jähriger Patient mit HOCM. Links: vor transkutaner Ablation der Septumhypertrophie (TASH), rechts nach TASH (1 Tag später). CKmax 700 U/l. Die ST-Hebungen in V1- bis V3 vor TASH sind nicht Ausdruck einer Ischämie. Nach TASH zeigt sich eine typische ST-Hebung in V1 und V2. Im Rahmen der Intervention neu entstandener Rechtsschenkelblock (die QRS-Dauer hat von 100 ms auf 140 ms zugenommen). Nach TASH stärker ausgeprägte Linksabweichung der QRS-Achse.
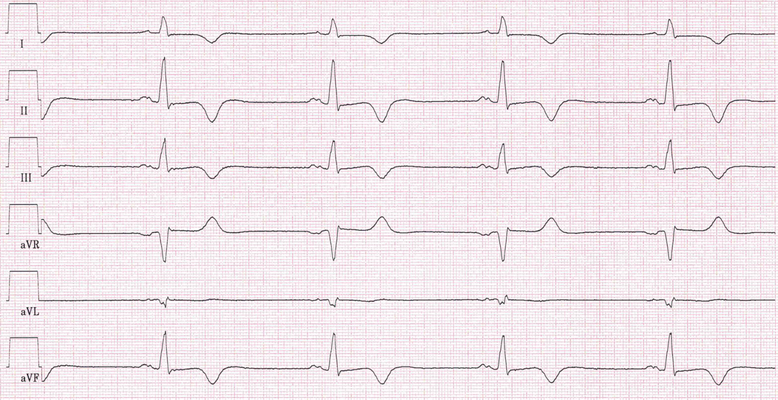
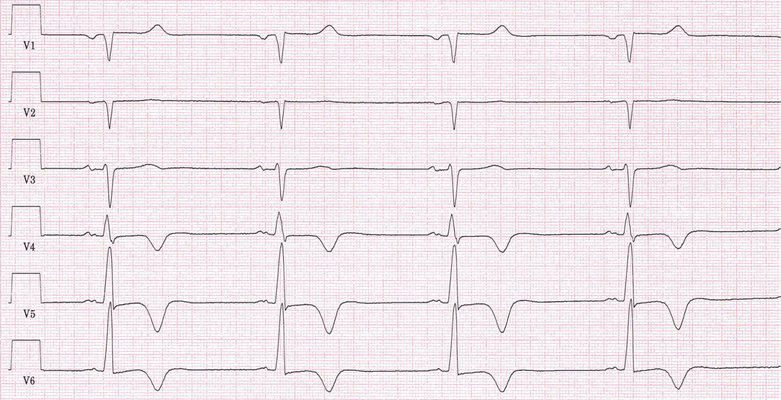
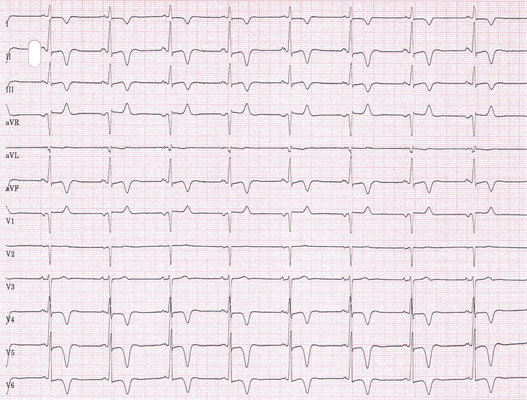
Abb.: 57-jähriger Patient mit einer hypertrophen Kardiomyopathie mit apikal lokalisierter Hypertrophie (apikale HCM). Typisch sind tief-negative T-Wellen linkspräkordial. Linsk: Extremitätenableitungen, 50 mm/s; Mitte: Brustwandableitungen, 50 m/s; Rechts: 12-Kanal-EKG, 25 mmHg.
Spontane Arrhythmien
Bei Patientinnen und Patienten mit HCM besteht eine deutlich erhöhte Neigung zu Rhythmusstörungen. Während nicht-anhaltende Kammertachykardien (nsVT) relativ häufig auftreten – sie werden bei etwa 25 % der Betroffenen nachgewiesen –, sind anhaltende Kammertachykardien vergleichsweise selten dokumentiert.
Ein Langzeit-EKG über mindestens 48 Stunden gehört zu den Basisuntersuchungen bei HCM, da sich Arrhythmien aufgrund ihrer hohen Spontanvariabilität nur durch längerfristiges Monitoring zuverlässig erfassen lassen.
Die HCM zählt zu den häufigsten Ursachen für plötzliche Herztodesfälle bei jungen Menschen, insbesondere bei Sportlern. Als zugrunde liegendes Arrhythmiesubstrat gelten die im hypertrophierten Myokard chaotisch angeordneten Herzmuskelfasern (myocardial disarray), häufig begleitet von Myokardfibrose.
Auch Vorhofflimmern und andere atriale Tachyarrhythmien treten bei HCM gehäuft auf. Das erstmalige Auftreten von Vorhofflimmern kann dabei ein Hinweis auf eine Krankheitsprogression sein.

Abb.: 71-jährige Patienten mit HOCM. Anhaltende Kammertachykardie (137/min). Negative QRS-Komplexe in den Einthovenableitungen (Nord-West-Achse von QRS, "no man´s land"). Rechtsschenkelblock.
Plötzlichen Herztod: Kalkulation des Risikos
Die HCM ist eine der häufigsten Ursachen für plötzliche Todesfälle in jungem Lebensalter. Bei allen betroffenen muss diesbezüglich eine Risikostratifizierung erfolgen. Im Internet ist ein Kalkulator verfügbar, mit dessen Hilfe sich das Risiko für das Auftreten eines plötzlichen Herztodes berechnen bzw. abschätzen lässt (Abb.). Zu den klassischen Risikofaktoren gehören unter anderem nicht-anhaltende Kammertachykardien und der familiäre plötzliche Herztod. Die Bedeutung neuer Parameter (z. B. Fibrose in der Magnet-Resonanz-Tomographie) für die Risikostartifizierung ist Gegenstand aktueller Diskussionen und Untersuchungen.

Abb.: Internat-basierter Kalkulator zur Bestimmung des Risikos bei Vorliegen einer hypertrophen Kardiomyopathie innerhalb der nächsten 5 Jahre an einem plötzlichen Herztod zu versterben.
Abgafragt werden konventionelle und relativ neue Risikofaktopren für ein plötzliches Versterben bei HCM.
Diagnostische Wertigkeit des EKGs und Differenzialdiagnosen
Die meisten Patienten mit bedeutsamer Hypertrophie weisen auch EKG-Veränderungen auf. Insofern ist das EKG bei HCM ein sehr sensitives diagnostisches Verfahren. Die
Spezifität ist eingeschränkt, andere Ursachen für die auftretenden EKG-Veränderungen müssen abgeklärt werden. Das EKG gehört, zusammen mit der Echokardiographie, zu den im Rahmen eines
Familienscreenings durchgeführten Basisuntersuchungen. Zu den wesentlichen Differenzialdiagnosen gehören die arterielle Hypertonie und myokardiale
Speichererkrankungen (z. B. Morbus Fabry). An letztere sollte insbesondere dann gedacht werden, wenn eine
periphere Niedervoltage vorliegt.
Literatur (frei zugänglich im Internet)
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J 2014;35:2733-2779.
- Rowin EJ, Maron MS. The Role of Cardiac MRI in the Diagnosis and Risk Stratification of Hypertrophic Cardiomyopathy. Arrhythmia & Electrophysiology Review. 2016;5(3):197-202.
Links
- Calculator für die Berechnung des Risikos für einen plötzlichen Herztod (Europäische Gesellschaft für Kardiologie).
- Orphanet (Datenbank zu seltenen Krankheiten und Medikamenten zur Behandlung seltener Krankheiten; wird unter französischer Federführung mit Förderung durch die Europäische Union betrieben).
Assoziierte Seiten