Long-QT-Syndrom
Das angeborene Long-QT-Syndrom ist durch eine Verlängerung der myokardialen Repolarisation (QT/QTc-Intervall-Verlängerung im Oberflächen-EKG) charakterisiert, die mit einer erhöhten Neigung zum Auftreten maligner Arrhythmien vom Typ der Torsade de Pointes einhergeht. Zu Rhythmusstörungen kommt es insbesondere bei verstärkter adrenerger Stimulation (körperlicher und/oder psychischer Belastung). Da die Rhythmusstörungen in der Regel spontan sistieren, stehen durch sie verursachte Synkopen klinisch im Vordergrund. Bei Degeneration von Torsade de Pointes in Kammerflimmern kann ein plötzlicher Herztod resultieren. Bislang sind mehr als 20 Gene bekannt, von denen angenommen wird, dass Mutationen krankheitsverursachend sein können. Die kodierten Proteine sind entweder Haupt- oder Untereinheiten von Ionenkanälen bzw. diese regulierende Proteine. Zu den am häufigsten betroffenen Genen gehören KCNQ1 (LQT1), KCNH2 (LQT2) und SCN5A (LQT3).
Diagnostischer Score nach Schwartz
Der nachfolgend aufgeführte Score stammt aus der Ära, in der noch keine genetische Testung möglich war (1993). Er berücksichtigt klinische und elektrokardiographische Kriterien. Die genetische Diagnostik ist heute bei Verdacht auf das Vorliegen eines Long-QT-Syndroms Routine. Allerdings dient sie eher der Diagnosesicherung als der Stellung der Diagnose, die weiterhin klinisch-elektrokardiographisch erfolgt. Zunächst wir der Index-Patient genetisch untersucht. Lässt sich eine Mutation identifizieren (in 80-90 % der Fälle), wird die engere Familie genetisch untersucht.
|
Parameter |
Punktwert |
Parameter |
Punktwert |
|
EKG-Veränderungen |
Eigenanamnese |
||
|
QTc > 480 ms |
3 |
Synkope, stressinduziert |
2 |
|
QTc 460–480 ms |
2 |
Synkope, andere Ursache |
1 |
|
QTc 450–459 ms (Mann) |
1 |
Innenohrschwerhörigkeit |
0,5 |
|
QTc > 480 ms (unter Belastung) |
1 |
|
|
|
Torsade de Pointes |
2 |
Familienanamnese |
|
|
T-Wellen-Alternans |
1 |
Long-QT-Syndrom in der Familie |
1 |
|
T-Wellen-Kerbungen |
1 |
Plötzlicher Herztod vor dem 30. Lebensjahr |
0,5 |
|
Niedrige Herzfrequenz |
0,5 |
|
|
Tab.: Score mit den diagnostischen Kriterien für ein angeborenes Long-QT-Syndrom. Hohe Wahrscheinlichkeit für ein Long-QT-Syndrom: >3,5 Punkte.; mittlere Wahrscheinlichkeit: 1,5 - 3 Punkte., geringe Wahrscheinlichkeit: < 1,5 Punkte.
Sekundäre Ursachen für eine QTc-Verlängerung müssen unbedingt ausgeschlossen werden. Zu den wichtigsten Ursachen für eine erworbene QT-Verlängerung (bei normaler QRS-Dauer) gehören eine Serum-Hypokaliämie sowie die Einnahme repolarisationsverlängernder Medikamente. Eine Liste der QT-verlängernden Medikamente ist im Anhang aufgeführt. Da auch neue Medikamente u. U. das QT-Intervall verlängern können, sei auf die Internet-Quelle www.torsades.org verwiesen, die ständig aktualisiert wird.
EKG
Traditionell wird von einer QTc-Verlängerung gesprochen, wenn das QTc-Intervall, berechnet anhand der Bazett-Formel 440 ms überschreitet. In der letzten Zeit werden vielfach geschlechtsabhängige Normwerte propagiert (> 450 für Männer, > 470 ms für Frauen). Die verwendeten Normalwerte für das QT-Intervall gelten nur bei normaler QRS-Dauer (<120 ms).Ein typisches LQT-EKG weist nicht nur eine abnorme Verlängerung von QTc, sondern auch Veränderungen der Morphologie der T-Welle auf (Abb.). In 20-25% der Long-QT-Fälle ist das QTc-Intervall normal. Solche Fälle können nur mittels molekulargenetischer Diagnostik identifiziert werden.
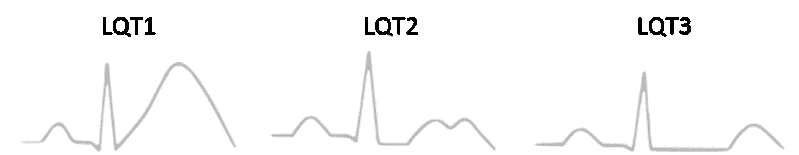
Abb.: Die einzelnen Unterformen des Long-QT-Syndroms weisen elektrokardiographisch Unterschiede auf, die bei den ersten 3 Formen (LQT1-LQT3), die auch die häufigsten Formen repräsentieren, besonders ausgeprägt sind. Bei einem LQT1 imponiert eine breitbasige T-Welle mit relativ hoher Amplitude. Charakteristisch für ein LQT2 ist die Kerbung der T-Welle. Beim LQT3 findet sich ein relativ langes isoelektrisches ST-Segment, die T-Welle weist eine schmale Basis und eine relativ niedrige Amplitude auf.
EKG bei LQT1
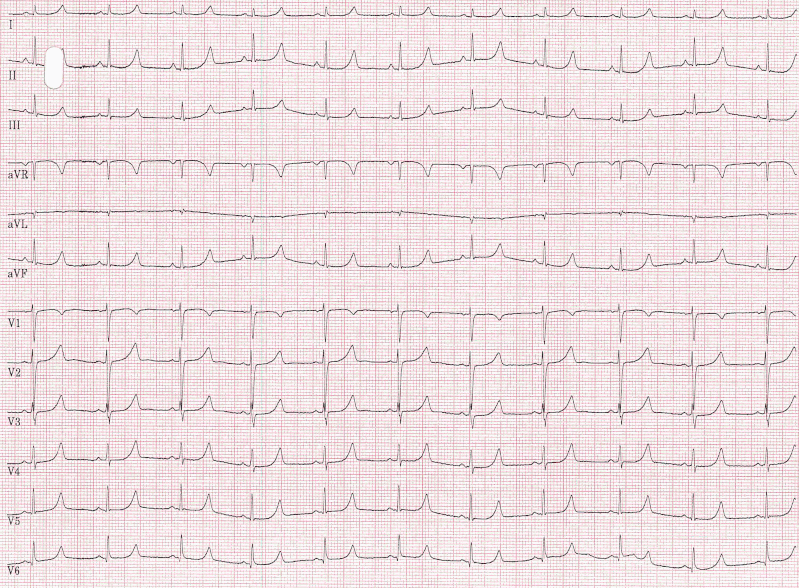
Abb.: 25-jährige Patientin mit Long-QT-Syndrom (LQT1). Herzfrequenz: 65 Schläge/min, QT: 466 ms, QTc(Bazett): 484 ms. Das relativ lange isoelektrische ST-Segment lässt an ein LQT3 denken, das aber bei der genetischen Untersuchung ausgeschlossen werden konnte.
EKG bei LQT2

Abb.: 27-jährige Patient mit Long-QT-Syndrom (LQT2). Herzfrequenz: 68 Schläge/min, QT: 516 ms, QTc(Bazett): 548 ms. Besonders charakteristisch sind die Kerbungen der T-Welle in den Ableitungen II, III, aVF sowie V2 und V3.

Abb.: 26-jähriger Patient mit einem Long-QT-Syndrom (LQT2), bislang keine kardialen Ereignisse. Dargestellt sind repräsentative Schläge aller 12-Ableitungen in Ruhe, unter Belastung und nach Belastung (Fahrradergometrie). Die Pfeile kennzeichnen gekerbte T-Wellen, die typisch für ein LQT2 sind. Das QT-Intervall ist deutlich verlängert (die T-Wellen sind so lang, dass sie bei der Darstellung durch das Gerät fast abgeschnitten wirken). Unter Belastung tritt eine QT-Verkürzung mit einer weitgehenden Normalisierung der Morphologie auf. Nach Belastung Wiederauftreten der T-Wellen-Kerbungen und der QT-Verlängerung. Bei Kindern sind Kerbungen von T bei normaler QT/QTc-Dauer ein physiologisches Phänomen.
EKG bei LQT3


Abb.: 24-jährige Patienten mit einem Long-QT-Syndrom (LQT3). Charakteristisch ist das lange isoelektrische ST-Segment und die schmalbasige T-Welle. Anklicken um zu vergrößern.

Abb.: Säugling mit einem angeborenen Long-QT-Syndrom (LQT3). Das QT-Intervall ist exzessiv verlängert. Die typische T-Wellen-Konfiguration (langes isoelektrisches Element, schmalbasige T-Welle) ist besonders gut in den Brustwandableitungen sichtbar.
Spontane Arrhythmien
Die für ein angeborenes langes QT-Syndrom charakteristische Rhythmusstörung ist Torsade de Pointes. Die Arrhythmie tritt bevorzugt unter Belastung auf und endet meistens spontan; sie kann aber auch in Kammerflimmern degenerieren und so zu einem plötzlichen Herztod führen.
Diagnostischer Stellenwert des EKGs bei langem QT-Syndrom
Die abnorme QT-Verlängerung ist pathognomonisch für das Long-QT-Syndrom. Für die Diagnosestellung ist der Ausschluss sekundärer Ursachen für eine QT/QTc-Verlängerung notwendig (Hypokaliämie, Hypocalcämie, Medikamente). Eine normale QT/QTc-Dauer schließt das Vorliegen eines langen QT-Syndroms nicht aus, daher erfolgt auch bei einem normalen QT/QTc-Intervall eine genetische Diagnostik, wenn die Familie betroffen ist. Die Dauer des QT/QTc-Intervalls ist sehr variabel, daher sollten auch im Zusammenhang mit einer Familienabklärung immer mehrere EKGs (zu unterschiedlichen Zeitpunkten, z. B. in Abständen von Wochen) registriert werden.
Literatur
- Giudicessi JR, Ackerman MJ. Genotype- and Phenotype-Guided Management of Congenital Long QT Syndrome. Current Probl Cardiol 2013;38:417-455.
- Schulze-Bahr E, Fenge H, W Haverkamp, et al. Long QT syndrome and life threatening arrhythmia in a newborn: molecular diagnosis and treatment response. Heart. 2004 Jan; 90(1): 13–16.
- Silvia G. Priori, Arthur A. Wilde, Minoru Horie, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013.
- Schulze-Bahr E, Klaassen S, Abdul-Khaliq H. Gendiagnostik bei kardiovaskulären Erkrankungen – Positionspapier der Deutschen Gesellschaft für Kardiologie (DGK) und der Deutschen Gesellschaft für Pädiatrische Kardiologie (DGPK). Kardiologe 2015;9:213–243.
Links
- www.torsades.org (bzw. www.credibledrugs.org): Informationen für Ärzte und Patienten zu Medikamenten, die zu vermeiden sind.
- Long-QT-Syndrom bei Orphanet (Datenbank zu seltenen Krankheiten und Medikamenten zur Behandlung seltener Krankheiten; wird unter französischer Federführung mit Förderung durch die Europäische Union betrieben).
- Long-QT-Syndrom: Informationen für Patienten (kardiologie-spreebogen.de)